

Bác sĩ: ThS.BS Dương Thị Thuỷ
Chuyên khoa: Nhi khoa
Năm kinh nghiệm: 15 năm
Đa u tủy (MM) thường được đặc trưng bởi sự tăng sinh tân sinh của các tế bào huyết tương tạo ra một globulin miễn dịch đơn dòng. Các tế bào huyết tương tăng sinh trong tủy xương và có thể dẫn đến phá hủy bộ xương trên diện rộng với các tổn thương tiêu xương, chứng loãng xương và / hoặc gãy xương bệnh lý.
Đa u tủy xương là bệnh ung thư khá phổ biến hiện nay.
MM là một bệnh ung thư tương đối phổ biến, chiếm khoảng 1-2% tổng số các bệnh ung thư và hơn 17% các khối u ác tính huyết học. Bệnh này phổ biến ở nam giới hơn phụ nữ và phổ biến hơn ở những người gốc Phi. Đây là một căn bệnh không thể chữa khỏi và là nguyên nhân của khoảng 20% trường hợp tử vong do bệnh ác tính huyết học và 2% trường hợp tử vong do tất cả các bệnh ung thư nói chung.
Tỷ lệ mắc hàng năm
- Dữ liệu từ cơ quan đăng ký Giám sát, Dịch tễ học và Kết quả Cuối cùng ( SEER ) của Hoa Kỳ ước tính 32.000 trường hợp mới mắc MM và 13.000 trường hợp tử vong do MM hàng năm ở Mỹ. Điều này tương quan với tỷ lệ mắc hàng năm khoảng 7 trên 100.000 nam và nữ mỗi năm. Một tỷ lệ mắc bệnh tương tự đã được báo cáo ở Canada, khu vực Nam Thames của Vương quốc Anh, và ở châu Âu nói chung. Trên toàn thế giới, có khoảng 180.000 trường hợp mắc và 117.000 trường hợp tử vong mỗi năm do MM.
Trong khi một số báo cáo cho thấy tỷ lệ mắc bệnh tăng lên theo thời gian, điều này có thể phản ánh việc gia tăng sử dụng các xét nghiệm thông thường trong phòng thí nghiệm, nâng cao nhận thức về MM và nâng cao tính sẵn có và sử dụng các cơ sở y tế, đặc biệt là người lớn tuổi.
Phân bố theo tuổi và giới tính
- MM phần lớn là bệnh của người lớn tuổi. Tuổi trung bình khi chẩn đoán là 65 đến 74 tuổi; tương ứng chỉ có 10% và 2% bệnh nhân dưới 50 và 40 tuổi. MM cũng thường xuyên hơn ở nam một chút so với nữ (khoảng 1,4: 1).
Sự thay đổi theo sắc tộc
- MM xảy ra ở mọi chủng tộc và mọi vị trí địa lý. Tỷ lệ mắc bệnh khác nhau tùy theo dân tộc; tỷ lệ mắc bệnh ở người Mỹ gốc Phi và người da đen cao gấp hai đến ba lần ở người da trắng trong các nghiên cứu từ Hoa Kỳ và Vương quốc Anh
Yếu tố nguy cơ
- Nguy cơ mắc MM tăng theo chỉ số khối cơ thể. Ngoài ra còn có mối liên quan giữa phơi nhiễm chất độc da cam và MM. Dữ liệu cũng cho thấy rằng trong số những bệnh nhân mắc bệnh gammopathy đơn dòng có ý nghĩa chưa xác định (MGUS), có sự gia tăng nguy cơ tiến triển thành MM ở những người đã tiếp xúc với chất độc da cam.
Nguy cơ gia đình
- Một phần nhỏ các trường hợp có tính chất gia đình với ước tính khoảng 3 trường hợp gia đình trên 1000 bệnh nhân mắc MM. Nguy cơ phát triển MM cao hơn khoảng 3,7 lần đối với những người có mức độ đầu tiên tương đối với MM.
MM được cho là phát triển từ giai đoạn tiền ác tính không có triệu chứng của sự tăng sinh tế bào huyết tương vô tính được gọi là bệnh gammopnal đơn dòng không xác định (MGUS). MGUS hiện diện trên 3% dân số trên 50 tuổi, và tiến triển thành u tủy hoặc bệnh ác tính liên quan với tỷ lệ 1% mỗi năm.
Trong khi MGUS không có triệu chứng, MM được đặc trưng bởi tổn thương cơ quan cuối, bao gồm tăng calci huyết, rối loạn chức năng thận, thiếu máu hoặc tổn thương xương lytic. Ở một số bệnh nhân, có giai đoạn không có triệu chứng sau tiến triển dần thành tiền ác tính trung gian được gọi là đa u tủy âm ỉ (SMM) có thể được nhận biết trên lâm sàng. Những bệnh nhân này có thể đã được chẩn đoán khi đang tiến triển từ MGUS thành MM, hoặc có thể đại diện cho MGUS sinh học với số lượng MGUS cao hơn.
Các yếu tố nguy cơ liên quan đến bệnh nhân và môi trường đối với MGUS đã được đề xuất, nhưng nguyên nhân chính xác của sự phát triển MGUS vẫn còn khó nắm bắt.
Các mô hình tương tự của một số bất thường di truyền tế bào nhất định có thể được tìm thấy trong các tế bào huyết tương vô tính của MGUS và MM. Những bất thường về di truyền tế bào này được cho là dẫn đến việc tạo ra một dòng tế bào plasma. Mặc dù sự kiện dẫn đến những thay đổi di truyền này có thể khác nhau, nhưng quá trình có thể xảy ra nhất là phản ứng bất thường của tế bào huyết tương đối với kích thích kháng nguyên.
Cơ chế bệnh học của MM là một quá trình phức tạp dẫn đến sự sao chép của một dòng nhân bản ác tính có nguồn gốc tế bào huyết tương. Trong khi một số bước trong lộ trình này đã được làm sáng tỏ. Hầu như tất cả các trường hợp MM đều có trước bởi rối loạn tăng sinh tế bào huyết tương tiền ác tính được gọi là bệnh gammopnal đơn dòng chưa xác định (MGUS). MGUS hiện diện trên 3% dân số trên 50 tuổi, và tiến triển thành u tủy hoặc bệnh ác tính liên quan với tỷ lệ 1% mỗi năm.
Cơ chế bệnh học của MM là một quá trình phức tạp dẫn đến sự sao chép của một dòng nhân bản ác tính có nguồn gốc tế bào huyết tương
Cơ chế bệnh sinh của MM có thể được khái niệm là hai quá trình tuần tự:
Sự hình thành của MGUS
- MGUS dường như phát triển do kết quả của các bất thường di truyền tế bào, nhiều trong số đó được cho là sản phẩm của phản ứng bất thường của tế bào huyết tương đối với kích thích kháng nguyên. Kết quả là một dòng tế bào huyết tương tạo ra globulin miễn dịch đơn dòng.
Tiến triển từ MGUS thành MM
- Tác động thêm đến dòng tế bào huyết tương, thông qua các bất thường di truyền bổ sung hoặc những thay đổi trong vi môi trường tủy xương, dẫn đến sự tiến triển của MGUS thành MM.
Các yếu tố nguy cơ liên quan đến bệnh nhân và môi trường đối với MGUS đã được đề xuất, nhưng nguyên nhân chính xác của sự phát triển MGUS vẫn còn khó nắm bắt.
Các mô hình tương tự của một số bất thường di truyền tế bào nhất định có thể được tìm thấy trong các tế bào huyết tương vô tính của MGUS và MM. Những bất thường về di truyền tế bào này được cho là dẫn đến việc tạo ra một dòng tế bào plasma. Mặc dù sự kiện dẫn đến những thay đổi di truyền này có thể khác nhau, nhưng quá trình có thể xảy ra nhất là phản ứng bất thường của tế bào huyết tương đối với kích thích kháng nguyên.
Yếu tố nguy cơ
- Dữ liệu dịch tễ học cho thấy khuynh hướng di truyền cũng như các yếu tố nguy cơ tiềm ẩn khác bao gồm tuổi già, ức chế miễn dịch và tiếp xúc với môi trường. Các yếu tố nội tiết tố có thể đóng một vai trò nào đó, vì phụ nữ có tỷ lệ lưu hành theo độ tuổi thấp hơn đáng kể so với nam giới.
Một khuynh hướng di truyền chủ yếu được hỗ trợ bởi những phát hiện rằng tỷ lệ mắc MGUS thay đổi theo dân tộc và một phần nhỏ, nhưng không rõ, là gia đình. Những trường hợp như vậy có thể do gen chung hoặc do yếu tố môi trường.
Tiếp xúc với bức xạ, benzen, và các dung môi hữu cơ khác, thuốc diệt cỏ và thuốc diệt côn trùng cũng có thể đóng một vai trò nào đó. Tuy nhiên, số trường hợp được báo cáo cho mỗi yếu tố nguy cơ này là nhỏ.
Bất thường di truyền tế bào
- Các bất thường di truyền tế bào nguyên phát dường như đóng một vai trò chính trong sự phát triển của MGUS. Hầu hết, nếu không phải tất cả, các trường hợp MGUS và MM đều có bất thường về nhiễm sắc thể có thể được phát hiện bằng phương pháp lai huỳnh quang tại chỗ (FISH), tạo mẫu phổ đa sắc, lai bộ gen so sánh hoặc lập hồ sơ biểu hiện gen. Tỷ lệ các trường hợp chứng tỏ mỗi bất thường khác nhau tùy theo phương pháp phát hiện được sử dụng và giai đoạn bệnh. Hầu hết các trường hợp MGUS dường như được khởi phát cùng với sự kiện chuyển vị liên quan đến vị trí chuỗi nặng globulin miễn dịch (IgH) (khoảng 40%) hoặc sự không ổn định di truyền biểu hiện bằng trisomies (khoảng 40%) hoặc cả chuyển vị và trisomies (khoảng 10%).
Đáp ứng không bình thường đối với kích thích kháng nguyên
- Các sự kiện gây ra các bất thường di truyền tế bào được mô tả ở trên không được biết, nhưng người ta cảm thấy rằng yếu tố kích hoạt những thay đổi đó có liên quan đến kích thích kháng nguyên. Bản chất của kích thích kháng nguyên chưa được biết rõ và có thể khác nhau giữa các trường hợp.
Vì không phải tất cả các bệnh nhân mắc bệnh gammopathy đơn dòng chưa xác định (MGUS) đều phát triển MM, những thay đổi di truyền ban đầu dẫn đến MGUS là cần thiết nhưng không đủ cho sự phát triển của MM. MGUS tiến triển thành MM có triệu chứng với tỷ lệ nhất quán hàng năm cho thấy rằng sự tiến triển này có thể được giải thích bằng mô hình "đánh thứ hai ngẫu nhiên". Nguy cơ tiến triển là tương tự nhau bất kể thời gian đã biết của MGUS tiền căn, cho thấy rằng tác động thứ hai gây ra sự tiến triển là một sự kiện ngẫu nhiên.
Một số sự kiện tiến triển có thể đóng vai trò thúc đẩy này dẫn đến tăng hình thành khối u:
- Các thay đổi di truyền bổ sung (ví dụ: đột biến Ras, metyl hóa p16, đột biến p53)
- Tăng sinh tế bào do rối loạn điều hòa chu kỳ tế bào
- Loại bỏ tế bào chết theo chương trình (apoptosis)
- Những thay đổi trong vi môi trường tủy xương
Bệnh học của tổn thương cơ quan cuối
- Khi quần thể tế bào huyết tương vô tính được tạo ra và tiến triển thành MM, bệnh nhân xuất hiện các triệu chứng (ví dụ: tăng canxi huyết, tổn thương xương ly giải, rối loạn chức năng thận và thiếu máu) liên quan đến sự xâm nhập của tế bào huyết tương vào xương hoặc các cơ quan khác hoặc tổn thương thận.
Hầu hết bệnh nhân bị MM đều có các dấu hiệu hoặc triệu chứng liên quan đến sự xâm nhập của các tế bào huyết tương vào xương hoặc các cơ quan khác hoặc tổn thương thận do lắng đọng immunoglobulin. Trong khi biểu hiện lâm sàng thường là bán cấp tính, một tỷ lệ nhỏ bệnh nhân có biểu hiện cấp tính với những phát hiện cần được quan tâm và can thiệp nhanh chóng (ví dụ: chèn ép tủy sống, suy thận, tăng độ nhớt máu).
Triệu chứng và dấu hiệu của đa u tủy xương là thiếu máu và đau xương
Một phân tích hồi cứu trên 1027 bệnh nhân tuần tự được chẩn đoán mắc bệnh MM tại một cơ sở duy nhất đã tìm thấy các triệu chứng và dấu hiệu sau khi xuất hiện
- Thiếu máu: 73%
- Đau xương: 58%
- Tăng creatinine: 48%
- Mệt mỏi / suy nhược tổng thể: 32%
- Tăng calci huyết: 28%
- Giảm cân: 24%, một nửa trong số đó đã giảm được 9 kg
Các triệu chứng và dấu hiệu xuất hiện trong 5% hoặc ít hơn bao gồm: dị cảm (5%), gan to (4%), lách to (1%), nổi hạch (1%) và sốt (0,7%). Tràn dịch màng phổi và lan tỏa phổi do thâm nhiễm tế bào huyết tương rất hiếm và thường xảy ra ở bệnh tiến triển.
Thiếu máu: hiện diện ở 73% khi được chẩn đoán và 97% tại một số thời điểm trong suốt quá trình của bệnh. Thiếu máu thường dẫn đến tình trạng mệt mỏi và xanh xao khi khám sức khỏe.
Đau xương: Bệnh tiêu xương là một đặc điểm chính của bệnh MM có thể dẫn đến đau xương và gãy xương bệnh lý.
Trong cùng một nghiên cứu được mô tả ở trên, đau xương liên quan đến MM xuất hiện tại thời điểm chẩn đoán ở khoảng 60% bệnh nhân. Đau liên quan đến MM thường:
- Liên quan đến khung xương trung tâm (lưng, cổ, vai, xương chậu, hông) chứ không phải tứ chi
- Thường do cử động gây ra và ít gặp hơn vào ban đêm khi ngủ mặc dù có thể xảy ra khi thay đổi tư thế
- Cường độ thường nhẹ hoặc trung bình nhưng nặng lên đến 10 phần trăm.
Chiều cao của bệnh nhân có thể giảm vài inch vì xẹp đốt sống.
Bệnh thận: suy thận có thể là biểu hiện của MM.
Tăng calci huyết: Tăng calci huyết có thể xảy ra ở MM do quá trình khử khoáng xương do MM gây ra. Tăng calci huyết được tìm thấy ở 28% của một loạt bệnh nhân bị MM tại thời điểm chẩn đoán, nhưng đa số không có triệu chứng tăng canxi huyết.
Bệnh thần kinh: Mặc dù hiếm gặp, nhưng các phát hiện về thần kinh có thể yêu cầu đánh giá khẩn cấp về sự chèn ép tủy sống (liệt nửa người) hoặc tăng độ nhớt máu (chảy máu mũi, mờ mắt, các triệu chứng thần kinh, lú lẫn và / hoặc suy tim).
Biến chứng hay gặp: đau xương, gãy xương, chèn ép, tủy sống, suy thận, nhiễm trùng và huyết khối, di căn, tử vong.
Phân bố theo tuổi và giới tính
MM phần lớn là bệnh của người lớn tuổi. Tuổi trung bình khi chẩn đoán là 65 đến 74 tuổi; tương ứng chỉ có 10 và 2% bệnh nhân dưới 50 và 40 tuổi. MM cũng thường xuyên hơn ở nam một chút so với nữ (khoảng 1,4: 1).
Đa u tủy xương phần lớn là bệnh của người lớn tuổi.
Sự thay đổi theo sắc tộc
MM xảy ra ở mọi chủng tộc và mọi vị trí địa lý. Tỷ lệ mắc bệnh khác nhau tùy theo dân tộc; tỷ lệ mắc bệnh ở người Mỹ gốc Phi và người da đen cao gấp hai đến ba lần ở người da trắng trong các nghiên cứu từ Hoa Kỳ và Vương quốc Anh
Yếu tố nguy cơ
Nguy cơ mắc MM tăng theo chỉ số khối cơ thể. Ngoài ra còn có mối liên quan giữa phơi nhiễm chất độc da cam và MM. Dữ liệu cũng cho thấy rằng trong số những bệnh nhân mắc bệnh gammopathy đơn dòng có ý nghĩa chưa xác định (MGUS), có sự gia tăng nguy cơ tiến triển thành MM ở những người đã tiếp xúc với chất độc da cam. Tiếp xúc với bức xạ, benzen và các dung môi hữu cơ khác, thuốc diệt cỏ và thuốc diệt côn trùng cũng có thể đóng một vai trò nhỏ.
Nguy cơ gia đình
Một phần nhỏ các trường hợp có tính chất gia đình với ước tính khoảng 3 trường hợp gia đình trên 1000 bệnh nhân mắc MM. Nguy cơ phát triển MM cao hơn khoảng 3,7 lần đối với những người có mức độ đầu tiên tương đối với MM.
Hiện nay, bệnh đa u tủy xương không có phòng bệnh đặc hiệu.

Tập thể dục tránh thừa cân là biện pháp hữu hiệu trong việc phòng đa u tủy xương
Hạn chế dầu mỡ, tăng tập thể dục để tránh thừa cân. Hạn chế tiếp xúc ới bức xạ, benzen và các dung môi hữu cơ khác, thuốc diệt cỏ và thuốc diệt côn trùng.
Những trường hợp nhiễm chất độc da cam, có tiền sử gia đình cần thăm khám định kỳ, phát hiện bệnh sớm nếu có.
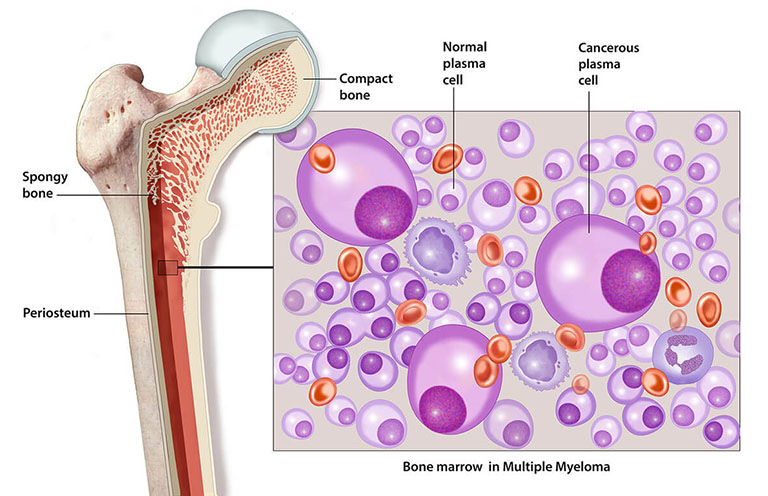
Tủy sống của bệnh nhân đa u tủy xương
Khi nào nghi ngờ MM
Việc chẩn đoán MM thường bị nghi ngờ do một (hoặc nhiều) biểu hiện lâm sàng sau:
- Đau xương với các tổn thương ly giải được phát hiện trên phim chụp xương thông thường hoặc các phương thức hình ảnh khác
- Tăng tổng nồng độ protein huyết thanh và / hoặc sự hiện diện của protein đơn dòng (M) trong huyết thanh hoặc nước tiểu
- Các dấu hiệu hoặc triệu chứng toàn thân gợi ý bệnh ác tính, chẳng hạn như thiếu máu không rõ nguyên nhân
- Tăng calci huyết, có triệu chứng hoặc được phát hiện tình cờ
- Suy thận cấp với phân tích nước tiểu hoặc hiếm khi có hội chứng thận hư do nhiễm amyloidosis nguyên phát đồng thời
Khi đánh giá một bệnh nhân nghi ngờ mắc MM, bệnh sử nên thu thập thông tin liên quan đến đau xương, các triệu chứng thiếu máu, các triệu chứng thần kinh và nhiễm trùng. Khám sức khỏe nên bao gồm khám thần kinh chi tiết.
Các xét nghiệm cận lâm sàng thông thường:
- Công thức máu đầy đủ và phân biệt với xét nghiệm lam máu ngoại vi.
- Sinh hóa: canxi huyết thanh, creatinine, albumin, lactate dehydrogenase (LDH) và beta-2 microglobulin (B2M).
- Tổng phân tích nước tiểu.
- Điện di protein huyết thanh (SPEP) với sự cố định miễn dịch và định lượng các globulin miễn dịch.
- Phân tích FLC huyết thanh
- Độ nhớt huyết thanh nên được đo nếu nồng độ protein M cao (nghĩa là> 5 g / dL) hoặc có các triệu chứng gợi ý tăng độ nhớt.
- Chọc hút và sinh thiết tủy xương với phương pháp định kiểu miễn dịch, di truyền tế bào thông thường và lai huỳnh quang tại chỗ (FISH).
- Đánh giá tủy xương được chỉ định cho tất cả bệnh nhân bị MM khi được chẩn đoán.
- Hình ảnh (CT, MRI, PET / CT) được ưu tiên hơn chụp X quang đơn thuần để phát hiện liên quan đến xương ở những bệnh nhân được đánh giá nghi ngờ MM. Một sự lựa chọn trong số này phụ thuộc vào tính khả dụng, chi phí, cơ sở điều trị và các đặc điểm lâm sàng.
Tiêu chuẩn chẩn đoán
Chẩn đoán MM yêu cầu đáp ứng các tiêu chí sau:
- Tế bào huyết tương tủy xương vô tính ≥10̀ hoặc u tế bào xương hoặc mô mềm đã được sinh thiết chứng minh: Tính vô tính nên được thiết lập bằng cách hiển thị hạn chế chuỗi ánh sáng kappa / lambda trên phép đo dòng chảy, hóa mô miễn dịch hoặc huỳnh quang miễn dịch. Phần trăm tế bào huyết tương tủy xương nên được ước tính từ mẫu sinh thiết. Khoảng 4% bệnh nhân có thể có ít hơn 10% tế bào huyết tương tủy xương vì sự tham gia của tủy có thể là khu trú, thay vì khuếch tán. Sinh thiết tủy xương lặp lại nên được xem xét ở những bệnh nhân như vậy.
Thêm một trong những điều sau:
- Sự hiện diện của cơ quan có liên quan hoặc suy mô:
+ Thiếu máu: Hemoglobin <10 g / dL (<100 g / L) hoặc> 2 g / dL (> 20 g / L) dưới mức bình thường.
+ Tăng calci huyết: Calci huyết thanh> 11 mg / dL (> 2,75 mmol / lít).
+ Suy thận: Độ thanh thải creatinin ước tính hoặc đo được <40 mL / phút hoặc creatinin huyết thanh> 2 mg/dL (177 micromol /lít).
+ Tổn thương xương: Một hoặc nhiều tổn thương tiêu xương có kích thước ≥5 mm trên chụp X quang xương, chụp cộng hưởng từ (MRI), chụp cắt lớp vi tính (CT), hoặc chụp cắt lớp phát xạ positron / chụp cắt lớp vi tính (PET / CT). Trong trường hợp không có tổn thương tiêu xương, những dấu hiệu sau đây không phải là dấu hiệu đầy đủ của tổn thương xương: tăng hấp thu fluorodeoxyglucose (FDG) trên PET, loãng xương, hoặc gãy chèn ép đốt sống. Khi nghi ngờ chẩn đoán, nên xem xét sinh thiết tổn thương xương.
- Sự hiện diện của một dấu ấn sinh học liên quan đến sự tiến triển gần như không thể tránh khỏi của tổn thương cơ quan cuối: Một hoặc nhiều trong số sau:
+ 60% tế bào huyết tương vô tính trong tủy xương.
+ Tỷ lệ FLC tham gia / không tham gia từ 100 trở lên (với điều kiện mức FLC liên quan ít nhất là 100 mg / L).
+ MRI với nhiều hơn một tổn thương khu trú (liên quan đến xương hoặc tủy xương)
Phân tầng nguy cơ:
- U tủy có nguy cơ cao: ít nhất một trong các tiêu chí lâm sàng hoặc bệnh lý sau đây là có MM nguy cơ cao:
+ t (4; 14), t (14; 16), t (14; 20), del17p13
+ Mức lactate dehydrogenase (LDH) ≥ 2 lần giới hạn bình thường
+ Đặc điểm của bệnh bạch cầu tế bào huyết tương nguyên phát (được xác định bởi ≥ 2000 tế bào huyết tương / microL máu ngoại vi hoặc ≥20% trên số đếm khác biệt bằng tay)
- U tủy có nguy cơ tiêu chuẩn: là những bệnh nhân thiếu tất cả các bất thường có nguy cơ cao được mô tả ở trên điều này bao gồm những bệnh nhân bị Trisomies, t (11; 14) và t (6; 14).
MM có thể điều trị song không thể chữa khỏi được. Điều trị thường được kết hợp giữa chống viêm steroid, hóa trị liệu, liệu pháp nhắm mục tiêu và ghép tế bào tự thân HCT và xạ trị đôi khi được sử dụng để giảm đau do tổn thương xương.
Đa u tủy xương có thể điều trị song không thể chữa khỏi được
Liệu pháp cảm ứng
Liệu pháp ban đầu cho bệnh nhân MM có triệu chứng phụ thuộc vào phân tầng nguy cơ, khả năng đủ điều kiện để cấy ghép tế bào tạo máu tự thân (HCT) và các nguồn lực sẵn có. Không có thỏa thuận chung nào về phác đồ khởi phát ưu tiên và các chuyên gia khác nhau sử dụng các phác đồ khác nhau.
Xác định tính đủ điều kiện cấy ghép
Tất cả bệnh nhân được đánh giá để xác định tính đủ điều kiện để cấy ghép tế bào tạo máu tự thân (HCT), điều này dường như kéo dài cả thời gian sống không biến cố và tổng thể khi so sánh với các chiến lược không cấy ghép.
Bệnh nhân đủ điều kiện cho HCT được điều trị cảm ứng từ ba đến bốn tháng trước khi thu thập tế bào gốc để giảm số lượng tế bào khối u trong tủy xương và máu ngoại vi, giảm triệu chứng và giảm tổn thương cơ quan cuối. Sau khi phục hồi từ việc thu thập tế bào gốc, bệnh
nhân có thể tiến hành trực tiếp đến HCT tự thân (chiến lược cấy ghép sớm) hoặc tiếp tục điều trị, thường với cùng một phác đồ được sử dụng để khởi phát, dự trữ HCT tự thân cho đến khi tái phát lần đầu (chiến lược cấy ghép muộn).
Cách tiếp cận điều trị trong u tủy nguy cơ tiêu chuẩn
- Bệnh nhân MM nguy cơ tiêu chuẩn đủ điều kiện cho HCT được điều trị bằng phác đồ bộ ba hoá chất trong ba đến bốn tháng trước khi lấy tế bào gốc.
- Sau khi thu thập tế bào gốc, nên làm HCT tự thân sớm hoặc muộn hơn là HCT đơn thuần hoặc HCT đồng sinh
- Những người tiến hành cấy ghép sớm được cung cấp ít nhất hai năm điều trị duy trì sau khi cấy ghép. Những người chọn trì hoãn HCT cho đến khi tái phát lần đầu tiên hoàn thành tổng cộng 8 đến 12 chu kỳ điều trị bộ ba, sau đó là duy trì dựa trên lenalidomide cho đến khi tái phát.
- Đối với hầu hết bệnh nhân không đủ điều kiện cho HCT, chúng tôi cung cấp phác đồ bộ ba dựa trên bortezomib trong 8 đến 12 chu kỳ, sau đó là điều trị duy trì dựa trên lenalidomide. Ở những bệnh nhân ốm yếu và không được cho là ứng cử viên của liệu pháp bộ ba, chúng tôi cung cấp liệu pháp bộ đôi với lenalidomide và dexamethasone liều thấp với sự chuyển đổi sang lenalidomide đơn tác nhân sau 9 chu kỳ.
U tủy có nguy cơ cao
Bệnh nhân MM có nguy cơ cao nên được khuyến khích đăng ký tham gia một thử nghiệm lâm sàng để điều tra các chiến lược điều trị mới, chúng kém với tất cả các lựa chọn điều trị thông thường. Đối với những bệnh nhân đủ điều kiện cấy ghép, có thể điều trị cảm ứng sau đó là HCT tự thân sớm và duy trì dựa trên chất ức chế proteasome (Lớp 2C). Những bệnh nhân không đủ điều kiện cấy ghép được cung cấp 8 đến 12 chu kỳ điều trị cảm ứng dựa trên bộ ba, sau đó là duy trì dựa trên chất ức chế proteasome.
Liệu pháp bổ sung
Ngoài liệu pháp hướng vào dòng nhân bản ác tính, việc quản lý hầu hết bệnh nhân bị MM bao gồm các biện pháp phòng ngừa để giảm tỷ lệ biến cố xương, tổn thương thận, nhiễm trùng và huyết khối. Bệnh nhân bị MM cũng có thể yêu cầu các can thiệp cụ thể để kiểm soát tình trạng tăng calci huyết, thiếu máu và bệnh thần kinh.
Đánh giá đáp ứng
Bệnh nhân nên được đánh giá trước mỗi chu kỳ điều trị để xác định xem bệnh của họ đang đáp ứng như thế nào với liệu pháp
Bệnh tái phát
Hầu hết tất cả bệnh nhân bị MM sẽ tái phát và cần điều trị thêm. MM tái phát hoặc kháng trị thường được xác định khi giám sát định kỳ. Các lựa chọn điều trị cho bệnh nhân bị bệnh MM tái phát hoặc khó chữa bao gồm HCT, điều trị lại phác đồ hóa trị trước đó hoặc thử nghiệm một phác đồ mới.
Multiple myeloma: Pathobiology - UpToDate
Multiple myeloma: Clinical features, laboratory manifestations, and diagnosis - UpToDate
Multiple myeloma: Overview of management - UpToDate

Bác sĩ: ThS.BS Dương Thị Thuỷ
Chuyên khoa: Nhi khoa
Năm kinh nghiệm: 15 năm
Quý khách hàng vui lòng lựa chọn dịch vụ y tế theo nhu cầu!


