

Bác sĩ: ThS.BS Trần Minh Dũng
Chuyên khoa: Tai mũi họng - Tai Mũi Họng
Năm kinh nghiệm: 9 năm
Liệt trên nhân tiến triển (Progressive Supranuclear Palsy - PSP) là một bệnh lý thần kinh thoái hóa hiếm gặp, tiến triển nhanh, thuộc nhóm Parkinson không điển hình (atypical parkinsonism). Bệnh đặc trưng bởi các triệu chứng như khó điều khiển ánh nhìn theo chiều dọc, mất thăng bằng, té ngã không rõ nguyên nhân, nói khó, nuốt khó, co cứng cơ và rối loạn hành vi – nhận thức liên quan đến vùng trán.
PSP xảy ra do sự tích tụ bất thường của một loại protein được gọi là “tau” trong tế bào thần kinh, tế bào hình sao và tế bào thần kinh đệm. Tình trạng này dẫn đến tổn thương ở nhiều vùng của não như hạch nền, thân não, tiểu não và vỏ não. Hình ảnh mô học đặc trưng bao gồm các đám rối sợi thần kinh hình cầu, chùm tế bào hình sao và các thể xoắn trong tế bào thần kinh đệm.
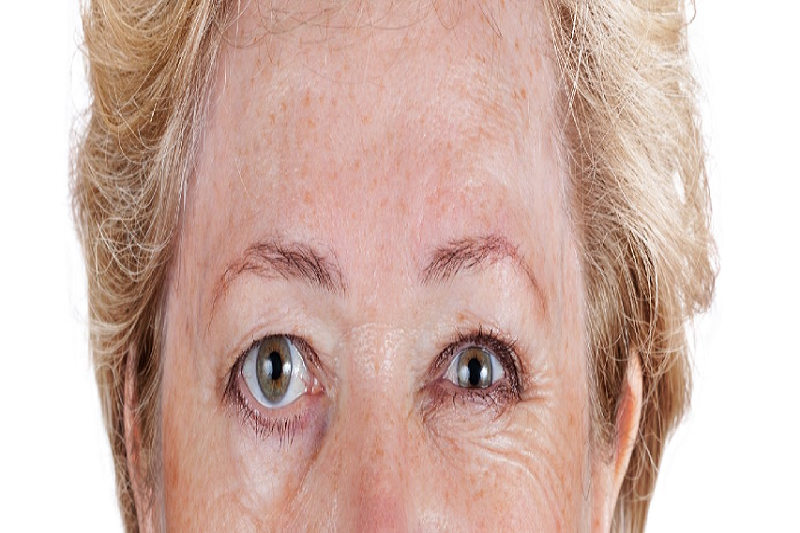
Liệt nhân trên tiến triển là một bệnh lý thoái hoá thần kinh hiếm gặp, tiến triển nhanh.
Phân loại lâm sàng
Theo tiêu chuẩn năm 2017 của Hiệp hội Parkinson và rối loạn vận động quốc tế (The International Parkinson and Movement Disorder Society - MDS), PSP có nhiều thể lâm sàng khác nhau, phản ánh các vị trí tổn thương trong não. Một số thể thường gặp gồm:
Các thể bệnh có thể chồng lấp và thay đổi theo thời gian. Khoảng 40% người bệnh chuyển từ thể này sang thể khác trong vòng bốn năm sau chẩn đoán.
PSP có diễn tiến nhanh và nặng. Hầu hết người bệnh mất khả năng tự chăm sóc trong vòng 3 - 4 năm từ khi khởi phát và tử vong trung bình sau 6 - 9 năm. Trong đó, thể điển hình PSP-RS thường tiến triển nhanh hơn các thể còn lại.
Liệt trên nhân tiến triển (PSP) là một bệnh lý thoái hóa thần kinh có liên quan đến sự tích tụ bất thường của protein tau. Dù cơ chế sinh bệnh chưa được hiểu rõ hoàn toàn, các nghiên cứu hiện nay cho thấy bệnh có thể liên quan đến sự kết hợp giữa tuổi tác, rối loạn chuyển hóa protein tau và yếu tố di truyền hoặc môi trường.
PSP thuộc nhóm bệnh do rối loạn protein tau. Ở người khỏe mạnh, protein tau giúp ổn định cấu trúc tế bào thần kinh. Khi bị rối loạn, protein tau bị biến đổi bất thường và tích tụ trong các tế bào thần kinh, tế bào hình sao và tế bào thần kinh đệm, hình thành các cấu trúc đặc trưng gồm:
Những tích tụ này gây chết tế bào, dẫn đến thoái hóa ở các vùng như hạch nền, thân não, tiểu não, vỏ trán và cả tủy sống. Hậu quả là người bệnh bị rối loạn vận động, mất thăng bằng, rối loạn điều khiển vận nhãn và suy giảm nhận thức
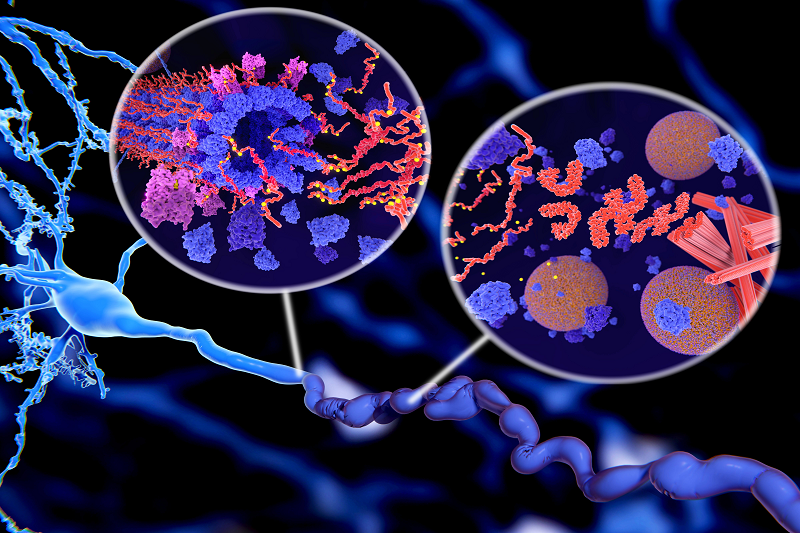
Liệt nhân trên tiến triển là bệnh lý thuộc nhóm rối loạn protein “tau”.
Một trong những hình ảnh đặc trưng của PSP là teo trung não, có thể nhận thấy trên phim cộng hưởng từ (MRI). Tổn thương tập trung nhiều nhất ở các vùng như nhân xám trung ương, nhân dưới đồi, nhân cầu nhạt trong, nhân điều khiển vận nhãn, vùng chất đen và vỏ não trước trán. Ngoài ra, thoái hóa tế bào thần kinh ở tủy sống cũng có thể góp phần gây rối loạn tiểu tiện.
Phần lớn trường hợp PSP không do di truyền. Tuy nhiên, một số ít bệnh nhân có tiền sử gia đình hoặc mang đột biến gen, trong đó đáng chú ý là đột biến MAPT – loại gen điều hòa sản xuất protein tau. Một số nghiên cứu cũng ghi nhận các biến thể di truyền như STX6, EIF2AK3, MOBP và nhóm con H1 của MAPT có thể làm tăng nguy cơ mắc bệnh. Dù vậy, những yếu tố này chỉ chiếm tỷ lệ rất nhỏ, đóng vai trò làm tăng nguy cơ chứ không phải là nguyên nhân trực tiếp gây bệnh.
Tuổi tác là yếu tố nguy cơ rõ ràng nhất – bệnh hầu như không xuất hiện trước 40 tuổi. Một số nghiên cứu còn cho thấy có thể có mối liên quan giữa PSP với:
Tuy nhiên, các yếu tố này hiện chỉ dừng lại ở mức độ quan sát và chưa được xác nhận là nguyên nhân rõ ràng gây bệnh. Trong tất cả các yếu tố, tuổi tác vẫn là yếu tố nguy cơ duy nhất được công nhận.

Tuổi tác là yếu tố nguy cơ rõ ràng nhất vì bệnh hầu như không xuất hiện trước 40 tuổi.
Một số biểu hiện thường gặp giúp nhận diện bệnh gồm:
Đặc điểm kinh điển trên khuôn mặt người bệnh là biểu hiện khuôn mặt cứng đờ, mắt mở to nhưng ít chớp mắt, khó biểu lộ cảm xúc – tạo nên vẻ mặt ngạc nhiên thường trực.
Tỷ lệ hiện mắc bệnh vào khoảng 5 - 7,1 trên 100.000 người. Tỷ lệ mới mắc mỗi năm là 1/100.000 và tăng rõ rệt theo tuổi, từ 1,7/100.000 ở độ tuổi 50 - 59 lên đến 14,7/100.000 ở nhóm tuổi 80- 89. Bệnh gặp ở cả nam và nữ với tỷ lệ tương đương, thường khởi phát trong độ tuổi cuối 60 đến đầu 70, hiếm khi xảy ra trước 40 tuổi.
Hiệp hội Parkinson và rối loạn vận động năm 2017 đưa ra hệ thống tiêu chuẩn chẩn đoán PSP, áp dụng cho tất cả các thể lâm sàng.
Tiêu chuẩn bắt buộc để chẩn đoán:
4 nhóm triệu chứng chính:
Mỗi biểu hiện trong bốn nhóm trên được phân loại theo mức độ nặng nhẹ, từ đó giúp xác định mức độ chẩn đoán: nghi ngờ, có thể, rất có thể hoặc xác định (chỉ được khẳng định sau khi giải phẫu bệnh).
Dù không có xét nghiệm đơn lẻ nào chẩn đoán chính xác PSP, một số kỹ thuật cận lâm sàng giúp củng cố chẩn đoán và loại trừ các nguyên nhân khác.
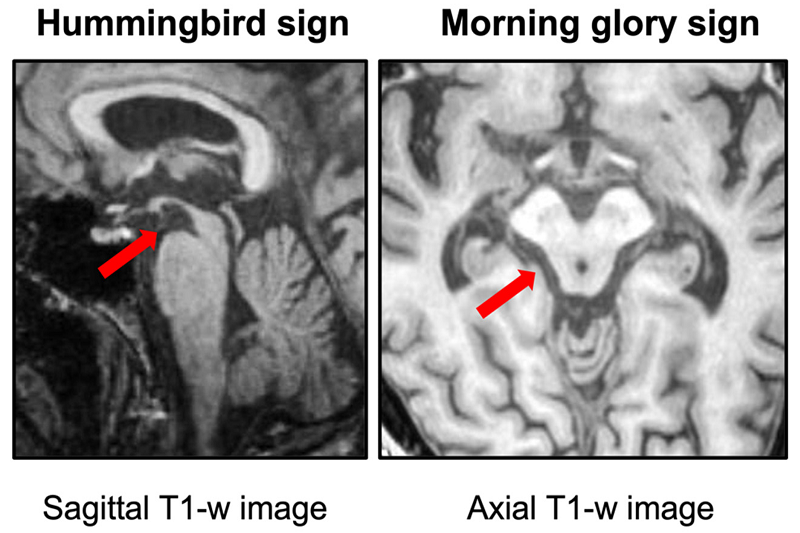
Dấu hiệu “chim ruồi” và “hoa sáng sớm” trên MRI não của người bệnh liệt nhân trên tiến triển.
Hiện nay chưa có phương pháp điều trị nào có thể làm chậm hoặc thay đổi tiến triển tự nhiên của bệnh liệt trên nhân tiến triển (PSP). Việc điều trị chủ yếu mang tính hỗ trợ, nhằm cải thiện chất lượng sống, duy trì khả năng vận động, hạn chế biến chứng và hỗ trợ tâm lý – xã hội cho người bệnh và người chăm sóc.

Người bệnh và gia đình nên trao đổi trước về việc lựa chọn nơi chăm sóc, an dưỡng cuối đời.
Một số thuốc hỗ trợ khác (chỉ nên cân nhắc từng trường hợp cụ thể):
Liệt trên nhân tiến triển là một bệnh lý thoái hóa thần kinh có diễn tiến nhanh và không thể phục hồi. Dù biểu hiện có thể khác nhau giữa các thể lâm sàng, phần lớn người bệnh đều suy giảm chức năng nghiêm trọng chỉ sau vài năm.
Hiện chưa có phương pháp điều trị nào có thể làm chậm hay đảo ngược tiến triển của bệnh. Mọi can thiệp hiện nay chỉ nhằm cải thiện triệu chứng và nâng cao chất lượng sống. Phần lớn bệnh nhân sẽ mất khả năng tự chăm sóc sau khoảng 3 đến 4 năm kể từ khi xuất hiện triệu chứng.
Thời gian sống trung bình sau khi được chẩn đoán là từ 6 đến 9 năm. Một số yếu tố cho thấy tiên lượng xấu gồm:
Tuổi khởi phát trên 70: Bệnh tiến triển nhanh hơn.
Thể PSP-RS: Nguy cơ tử vong sớm hơn thể PSP-P.
Suy giảm nhận thức hoặc rối loạn nuốt sớm: Dễ gặp biến chứng hơn.
Không đáp ứng với levodopa: Thường là thể bệnh nặng và tiến triển nhanh.
Việc đánh giá chính xác thể bệnh và theo dõi tiến triển bằng các công cụ khách quan (như thang điểm PSP-RS hoặc thiết bị đo dáng đi) có thể giúp tiên lượng tốt hơn và lựa chọn phương pháp chăm sóc phù hợp theo từng giai đoạn.
Tài liệu tham khảo:

Bác sĩ: ThS.BS Trần Minh Dũng
Chuyên khoa: Tai mũi họng - Tai Mũi Họng
Năm kinh nghiệm: 9 năm
Quý khách hàng vui lòng lựa chọn dịch vụ y tế theo nhu cầu!


