

Bác sĩ: ThS.BS Trần Minh Dũng
Chuyên khoa: Tai mũi họng - Tai Mũi Họng
Năm kinh nghiệm: 9 năm
Thalassemia là một nhóm bệnh lý huyết học di truyền, đặc trưng bởi tình trạng giảm hoặc mất khả năng tạo ra các chuỗi globin – là thành phần cấu tạo nên huyết sắc tố (hemoglobin). Khi quá trình này bị rối loạn, cơ thể không sản xuất đủ hemoglobin, dẫn đến tình trạng thiếu máu hồng cầu nhỏ nhược sắc mạn tính. Bệnh được chia thành hai nhóm chính: Alpha thalassemia là do giảm tổng hợp chuỗi alpha globin, còn beta thalassemia là do giảm tổng hợp chuỗi beta globin.
Alpha thalassemia thường xuất hiện ở người có nguồn gốc Đông Nam Á hoặc châu Phi, trong khi beta thalassemia phổ biến hơn ở các khu vực Địa Trung Hải, Trung Đông, Ấn Độ và Đông Nam Á. Tùy theo số lượng gen bị ảnh hưởng, bệnh có thể biểu hiện ở nhiều mức độ khác nhau: Từ thể nhẹ không có triệu chứng (người lành mang gen bệnh) đến thể nặng với biểu hiện rõ rệt như thiếu máu, lách to, biến dạng xương, chậm phát triển thể chất và nguy cơ quá tải sắt do truyền máu nhiều lần.

Thalassemia là một nhóm bệnh lý huyết học di truyền.
Tỷ lệ mắc bệnh có sự khác biệt theo khu vực địa lý. Ước tính có khoảng 5% dân số thế giới mang ít nhất một gen bệnh, và khoảng 900.000 người có biểu hiện lâm sàng rõ ràng trong thế kỷ 21, chủ yếu tập trung ở Trung Quốc, Ấn Độ và khu vực Đông Nam Á.
Thalassemia là bệnh di truyền xảy ra khi có các biến đổi bất thường tại hoặc gần cụm gen globin – những gen chịu trách nhiệm sản xuất chuỗi globin. Sự thay đổi này làm giảm hoặc mất hoàn toàn khả năng tổng hợp một loại chuỗi globin cụ thể, từ đó dẫn đến thiếu hụt hemoglobin. Hai dạng bệnh phổ biến là alpha và beta thalassemia, mỗi dạng có đặc điểm riêng về cơ chế hình thành và kiểu đột biến gây bệnh.
1. Nguyên nhân di truyền ở beta thalassemia
Đối với beta thalassemia, nguyên nhân là do các đột biến xảy ra tại gen HBB nằm trên nhiễm sắc thể số 11. Các đột biến này có thể làm mất hoàn toàn khả năng tạo chuỗi beta globin (gọi là đột biến β⁰), hoặc chỉ làm giảm một phần khả năng này (đột biến β⁺).
Những đột biến này có thể tác động đến nhiều quá trình: làm mất đoạn và tái tổ hợp bất thường trong cấu trúc gen, đột biến vùng khởi động (promoter) làm giảm phiên mã, gây sai lệch trong quá trình nối mRNA, mất codon khởi đầu hoặc tạo ra codon kết thúc sớm trong quá trình dịch mã, hoặc làm giảm độ ổn định của mRNA và chuỗi globin.
2. Nguyên nhân di truyền ở alpha thalassemia
Đối với alpha thalassemia, phần lớn trường hợp là do mất đoạn gen tại vùng HBA1 và HBA2 trên nhiễm sắc thể số 16. Khi một hoặc nhiều bản sao của gen bị mất, khả năng sản xuất chuỗi alpha bị suy giảm. Mức độ nghiêm trọng của bệnh phụ thuộc vào số lượng gen bị mất: mất 1 gen thường không gây triệu chứng; mất cả 4 gen sẽ dẫn đến tình trạng Hb Barts hoặc phù thai nặng, thường gây tử vong từ trong bụng mẹ. Ngoài ra, một số đột biến dạng điểm cũng có thể gây alpha thalassemia, như đột biến Hemoglobin Constant Spring làm giảm độ bền của mRNA, từ đó làm giảm tổng hợp chuỗi alpha.
3. Các yếu tố ngoài locus globin
Bên cạnh các tổn thương trong cụm gen globin, một số đột biến nằm ngoài vùng này cũng có thể gây ra biểu hiện giống thalassemia. Ví dụ, đột biến gen GATA1 làm giảm khả năng điều hòa biểu hiện của gen beta globin; hoặc đột biến gen ATRX có thể liên quan đến hội chứng alpha thalassemia kèm chậm phát triển tâm thần, hay thalassemia mắc phải ở người có rối loạn sinh tủy.
4. Cơ chế chung dẫn đến bệnh
Dù thuộc dạng alpha hay beta, điểm chung trong cơ chế bệnh sinh của thalassemia là sự mất cân bằng giữa chuỗi alpha và chuỗi beta trong quá trình tạo hemoglobin. Khi một loại chuỗi bị thiếu, chuỗi còn lại không bắt cặp được sẽ tích tụ và kết tủa trong tế bào hồng cầu. Điều này gây tổn thương màng tế bào, dẫn đến phá huỷ hồng cầu sớm ngay trong tủy xương (tạo máu không hiệu quả) hoặc khi ra ngoài máu (tán huyết). Chính sự tích tụ của các chuỗi globin dư thừa này là nguyên nhân gây ra nhiều biến chứng như biến dạng xương, gan lách to, sỏi mật và quá tải sắt.
Triệu chứng của thalassemia thay đổi tùy theo mức độ mất cân bằng giữa các chuỗi globin:
Thalassemia thể nhẹ thường được phát hiện tình cờ qua xét nghiệm máu với kết quả cho thấy hồng cầu nhỏ, nhược sắc.

Các xét nghiệm di truyền được thực hiện để xác định đột biến gen Thalassemia.
Việc điều trị thalassemia phụ thuộc vào thể bệnh và mức độ nặng, bao gồm các biện pháp hỗ trợ không dùng thuốc, điều trị bằng thuốc và các phương pháp can thiệp chuyên sâu như ghép tế bào gốc. Mục tiêu chính là kiểm soát tình trạng thiếu máu, phòng ngừa biến chứng do quá tải sắt và nâng cao chất lượng sống cho người bệnh.
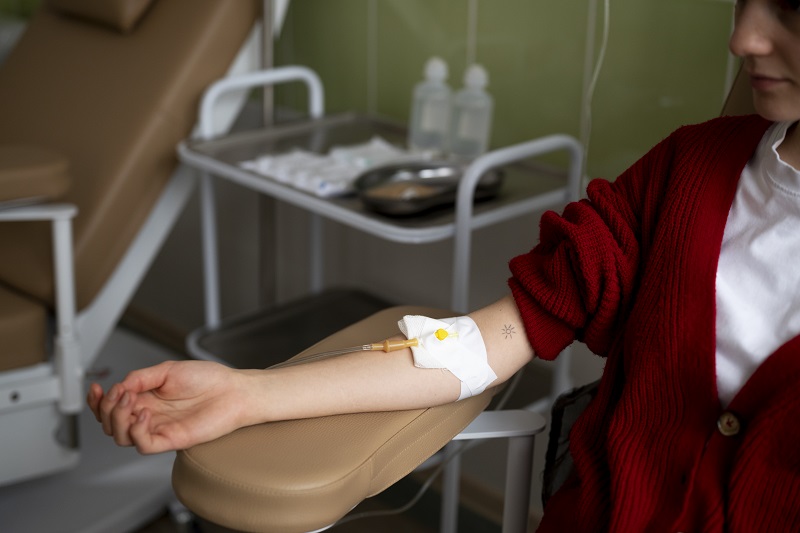
Truyền máu định kỳ là nền tảng trong điều trị Thalassemia thể nặng.
Tiên lượng của bệnh thalassemia phụ thuộc vào thể bệnh, mức độ thiếu máu, tần suất truyền máu, biến chứng do quá tải sắt và khả năng tiếp cận điều trị chuyên sâu như ghép tế bào gốc. Nhìn chung, bệnh càng nặng và nhu cầu truyền máu càng nhiều thì nguy cơ biến chứng càng cao nếu không được kiểm soát tốt.
Tài liệu tham khảo:

Bác sĩ: ThS.BS Trần Minh Dũng
Chuyên khoa: Tai mũi họng - Tai Mũi Họng
Năm kinh nghiệm: 9 năm
Quý khách hàng vui lòng lựa chọn dịch vụ y tế theo nhu cầu!


