

Bác sĩ: ThS.BS Trần Minh Dũng
Chuyên khoa: Tai mũi họng - Tai Mũi Họng
Năm kinh nghiệm: 9 năm
Bệnh bò điên (tên khoa học: Bovine Spongiform Encephalopathy – BSE) là một rối loạn thoái hóa thần kinh hiếm gặp ở bò. Bệnh không lây truyền thông qua đường tiếp xúc thông thường. Tuy nhiên, bệnh có thể lây từ động vật sang người qua đường tiêu hóa nếu ăn phải thịt bò bị nhiễm mầm bệnh. Khi xâm nhập vào cơ thể người, tác nhân gây bệnh có thể dẫn đến một biến thể hiếm gặp của bệnh Creutzfeldt-Jakob, gọi là variant CJD (vCJD) – một dạng bệnh lý não tiến triển, không thể chữa khỏi và luôn gây tử vong. Cả BSE và vCJD đều thuộc nhóm bệnh "não xốp truyền nhiễm" (transmissible spongiform encephalopathies – TSEs), với đặc trưng là sự tích tụ bất thường của prion – một loại protein sai cấu trúc có khả năng tự nhân bản và phá hủy mô thần kinh.
 Bệnh bò điên là một rối loạn thoái hóa thần kinh hiếm gặp ở bò, lây truyền sang người qua đường tiêu hóa.
Bệnh bò điên là một rối loạn thoái hóa thần kinh hiếm gặp ở bò, lây truyền sang người qua đường tiêu hóa.
Lịch sử phát hiện bệnh
BSE lần đầu tiên được phát hiện tại Anh vào năm 1986, nó nhanh chóng trở thành mối quan tâm toàn cầu khi hàng trăm nghìn con bò bị tiêu hủy để kiểm soát dịch. Đến năm 1995, những ca bệnh vCJD đầu tiên ở người được ghi nhận, chủ yếu tại Vương quốc Anh, sau khi người dân tiêu thụ các sản phẩm thịt có chứa mô thần kinh từ bò bệnh. Đặc biệt, nhiều bệnh nhân vCJD là người trẻ tuổi, điều này rất khác biệt so với thể CJD cổ điển thường gặp ở người cao tuổi. Sau khi xác định được mối liên hệ giữa BSE và vCJD, hàng loạt chính sách kiểm soát an toàn thực phẩm đã được ban hành ở nhiều quốc gia để ngăn chặn nguy cơ bùng phát đại dịch.
Ngoài vCJD ra, con người còn có thể mắc các thể CJD khác không liên quan đến bò điên, gồm:
Trong khi đó, vCJD được xem là hậu quả trực tiếp từ phơi nhiễm prion của BSE, thường xảy ra ở người trẻ và có diễn tiến bệnh chậm hơn so với thể CJD cổ điển.
Tính đến năm 2015, toàn cầu ghi nhận 229 ca vCJD, trong đó hơn 80% ở Vương quốc Anh. Ở các quốc gia khác như Pháp, Mỹ, Canada, số ca ghi nhận ở mức rất thấp. Mặc dù vCJD cực kỳ hiếm gặp, nhưng vì không thể điều trị và tỷ lệ tử vong 100% khiến căn bệnh này được xếp vào nhóm bệnh cần giám sát đặc biệt trong y học dự phòng. Một nghiên cứu tại Anh còn chỉ ra rằng khoảng 1/2.000 người có thể mang prion bất hoạt trong hệ bạch huyết, làm dấy lên lo ngại về khả năng tồn tại các ca bệnh tiềm ẩn.
Cả BSE ở bò và vCJD ở người đều xuất phát từ sự rối loạn cấu trúc của prion – một loại protein có trong hệ thần kinh. Bình thường, prion tồn tại ở dạng lành tính, nhưng khi bị biến đổi sang dạng bệnh lý, chúng không bị phân giải bởi enzym tiêu hóa, có khả năng tự nhân bản và tích tụ trong mô não, gây tổn thương thần kinh dạng xốp.
Prion bệnh lý không chỉ kháng lại các phương pháp tiệt trùng thông thường, mà còn không gây phản ứng miễn dịch, khiến cơ thể không có khả năng tự phát hiện và tiêu diệt chúng. Đây là điểm đặc biệt khiến prion khác biệt hoàn toàn với virus, vi khuẩn hay nấm.
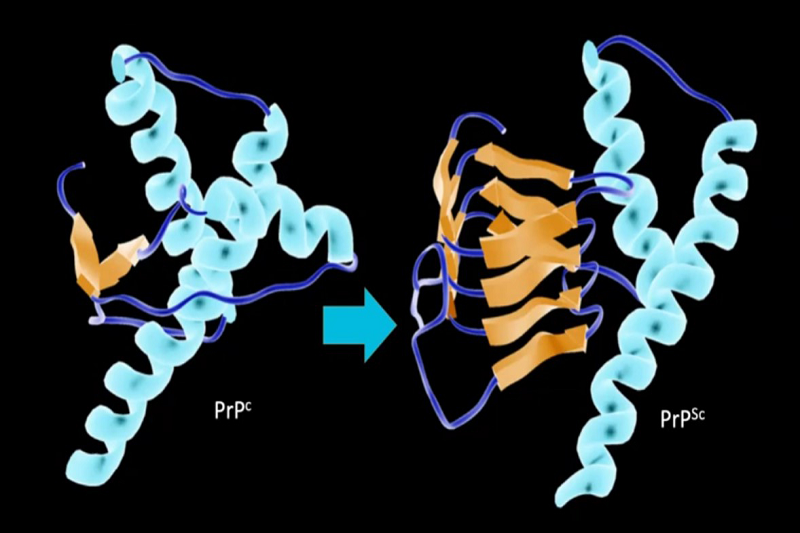 Nguyên nhân gây bệnh bắt nguồn từ rối loạn cấu trúc Prion - protein hệ thần kinh.
Nguyên nhân gây bệnh bắt nguồn từ rối loạn cấu trúc Prion - protein hệ thần kinh.
Các nghiên cứu dịch tễ học cho thấy dịch bệnh BSE bùng phát mạnh tại Anh vào những năm 1980-1990 có liên quan đến thức ăn chăn nuôi chế biến từ xác động vật – đặc biệt là xương, não và tủy sống của cừu và bò. Trong đó, prion từ cừu mắc bệnh Scrapie hoặc từ bò nhiễm BSE được cho là nguồn gốc ban đầu.
Việc loại bỏ bước xử lý nhiệt bằng dung môi trong quy trình chế biến thức ăn chăn nuôi đã vô tình làm prion không bị tiêu diệt, dẫn đến sự lây lan âm thầm qua nhiều thế hệ bò. Việc “biến bò – loài ăn cỏ – thành động vật ăn xác động vật” đã tạo điều kiện cho sự khuếch đại bất thường của prion trong quần thể gia súc, gây ra đại dịch BSE.
Khác với các thể CJD khác, vCJD là một bệnh lây từ động vật sang người. Con đường lây truyền chủ yếu là ăn phải các sản phẩm thịt chứa mô thần kinh (não, tủy sống, hạch bạch huyết…) của bò mắc BSE. Đây được gọi là lây truyền kiểu zoonotic (từ động vật sang người).
Sau khi prion bệnh lý xâm nhập qua đường tiêu hóa, chúng vượt qua niêm mạc ruột nhờ vào các tế bào M, rồi tập trung trong hệ bạch huyết như amidan, lách, ruột non… Cuối cùng, prion theo đường thần kinh thực vật (giao cảm và phó giao cảm) đến hệ thần kinh trung ương, gây tổn thương lan rộng và tiến triển không hồi phục.
Biểu hiện lâm sàng của vCJD có điểm đặc biệt là khởi phát âm thầm, dễ nhầm với các rối loạn tâm thần, đặc biệt ở người trẻ tuổi. Giai đoạn đầu, bệnh thường không rõ triệu chứng thần kinh rõ rệt mà biểu hiện qua thay đổi tính cách, cảm xúc thất thường và cảm giác khó chịu.
Các dấu hiệu thường gặp bao gồm:
Quá trình tiến triển của vCJD thường kéo dài hơn các thể CJD khác, với trung bình thời gian từ khi khởi phát đến lúc tử vong khoảng 14 tháng. Bệnh không đáp ứng điều trị và chỉ tiến triển liên tục đến tử vong.

vCJD là một bệnh lý thoái hóa thần kinh hiếm gặp nhưng có tiên lượng rất nghiêm trọng. Giống như các bệnh prion khác, vCJD luôn dẫn đến tử vong, và hiện vẫn chưa có phương pháp nào có thể đảo ngược hay làm chậm diễn tiến bệnh.
 Cho đến hiện tại, tỉ lệ tử vong của bệnh là 100%.
Cho đến hiện tại, tỉ lệ tử vong của bệnh là 100%.
Không có trường hợp nào ghi nhận hồi phục hoàn toàn từ vCJD. Tất cả bệnh nhân được chẩn đoán đều tiến triển không hồi phục, với tổn thương lan rộng ở hệ thần kinh trung ương. Việc điều trị chỉ mang tính giảm nhẹ triệu chứng và chăm sóc cuối đời.
Tuy nhiên, thời gian sống của bệnh nhân vCJD dài hơn so với các thể CJD cổ điển. Nếu như bệnh Creutzfeldt-Jakob tự phát có thể gây tử vong chỉ sau vài tháng, thì người mắc vCJD có thời gian sống trung bình khoảng 14 tháng kể từ lúc khởi phát triệu chứng. Một số trường hợp có thể kéo dài đến 24-36 tháng, tùy theo mức độ tổn thương, chất lượng chăm sóc và cơ địa người bệnh.
vCJD ảnh hưởng nghiêm trọng đến chất lượng sống cả về thể chất lẫn tinh thần:
Các biến chứng này không chỉ gây suy giảm chức năng sống mà còn tạo gánh nặng lớn cho người chăm sóc và hệ thống y tế.
Khác với các bệnh nhiễm trùng thông thường, vCJD không có khái niệm tái phát, vì nó là một bệnh tiến triển liên tục cho đến khi tử vong. Tuy nhiên, lo ngại lớn hiện nay là nguy cơ xuất hiện thêm ca bệnh mới từ những người mang prion tiềm ẩn mà chưa biểu hiện lâm sàng.
Một nghiên cứu giải phẫu bệnh trên hàng chục nghìn mẫu amidan và ruột thừa tại Anh cho thấy, tỷ lệ mang prion tiềm ẩn có thể lên tới 1/2.000 người, dù không có triệu chứng. Điều này làm dấy lên lo ngại rằng vCJD vẫn có thể tái xuất hiện trong tương lai, đặc biệt nếu các biện pháp kiểm soát prion trong thực phẩm và y tế bị lơ là.
Một số yếu tố có thể quyết định mức độ tiến triển và thời gian sống của bệnh nhân vCJD:
Vì thế, những người từng sống tại quốc gia có dịch BSE trước năm 1996 hoặc từng được ghép mô, truyền máu không rõ nguồn gốc sẽ không được hiến máu ở nhiều quốc gia như Hoa Kỳ.
vCJD thường xuất hiện ở người dưới 30 tuổi. Người trẻ có thể nhạy cảm hơn với prion do đáp ứng miễn dịch và cấu trúc thần kinh chưa lão hóa.
Các nhà khoa học phát hiện rằng, trong cơ thể mỗi người có một gen tên là PRNP, chịu trách nhiệm tạo ra loại protein gọi là prion. Ở vị trí số 129 trên gen này, mỗi người sẽ mang một trong ba kiểu di truyền: Met/Met, Met/Val hoặc Val/Val.
Thực tế, tất cả các trường hợp mắc bệnh vCJD được ghi nhận cho đến nay đều thuộc nhóm người có kiểu gen Met/Met. Điều này khiến các nhà khoa học cho rằng kiểu gen này có thể làm tăng nguy cơ mắc bệnh nếu bị phơi nhiễm prion từ thực phẩm nhiễm bệnh.
Lượng mô thần kinh nhiễm prion càng lớn thì nguy cơ mắc bệnh càng cao. Thịt xay, nội tạng hoặc các chế phẩm thịt chế biến không kỹ được xem là nhóm nguy cơ.
Một số trường hợp vCJD xảy ra sau khi bệnh nhân nhận truyền máu từ người hiến máu sau đó mắc vCJD. Điều này cho thấy prion có thể tồn tại trong máu, dù chưa có cách xét nghiệm máu phổ cập để sàng lọc.
Tổ chức Y tế Thế giới (WHO) và các trung tâm giám sát như CDC đưa ra tiêu chuẩn chẩn đoán vCJD dựa trên kết hợp giữa triệu chứng lâm sàng, hình ảnh học thần kinh và các xét nghiệm chuyên biệt.
Một số yếu tố gợi ý chẩn đoán vCJD:
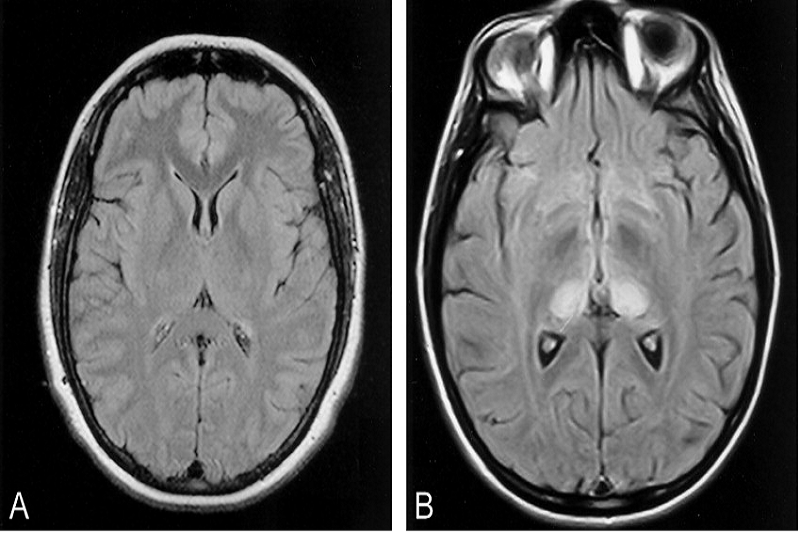 Dấu hiệu “Pulvinar sign” đặc trưng của bệnh trên MRI não.
Dấu hiệu “Pulvinar sign” đặc trưng của bệnh trên MRI não.
Hiện nay, không có phương pháp điều trị đặc hiệu nào có thể đảo ngược hay làm chậm tiến triển của bệnh vCJD. Tất cả các phương án chăm sóc đều mang tính hỗ trợ, nhằm duy trì chất lượng cuộc sống và giảm nhẹ triệu chứng trong giai đoạn bệnh tiến triển.
Mục tiêu chính của chăm sóc không dùng thuốc là giúp người bệnh cảm thấy thoải mái nhất có thể, đồng thời hỗ trợ người thân trong quá trình chăm sóc tại nhà hoặc cơ sở y tế.
Các biện pháp bao gồm:
Không có loại thuốc nào hiện tại được FDA phê duyệt để điều trị vCJD. Tuy nhiên, một số thuốc có thể giảm nhẹ triệu chứng:
Ngoài ra, thuốc như amantadine, memantine hoặc steroids từng được thử nghiệm nhưng không chứng minh được hiệu quả trong việc cải thiện tiến triển bệnh.
Mặc dù chưa có liệu pháp đặc trị, nhiều nghiên cứu thử nghiệm đang được tiến hành nhằm tìm ra giải pháp tiềm năng. Một số hướng tiếp cận đáng chú ý gồm:
Tóm lại, vCJD là bệnh lý thần kinh có tiên lượng rất xấu, gây tử vong không thể tránh khỏi sau thời gian tiến triển âm thầm. Dù số ca mắc hiện đã giảm mạnh nhờ kiểm soát thực phẩm và hệ thống giám sát y tế, việc duy trì cảnh giác, phát hiện sớm và chăm sóc giảm nhẹ vẫn là chiến lược then chốt để ứng phó với căn bệnh này.
Tài liệu tham khảo:

Bác sĩ: ThS.BS Trần Minh Dũng
Chuyên khoa: Tai mũi họng - Tai Mũi Họng
Năm kinh nghiệm: 9 năm
Quý khách hàng vui lòng lựa chọn dịch vụ y tế theo nhu cầu!


